La diagnosi di tumore cerebrale è un’esperienza profonda e travolgente che delinea “un prima e un dopo” nella vita di una persona. Dalla comparsa dei sintomi iniziali, passando per gli esami radiologici, fino alle valutazioni specialistiche, il percorso è carico di emozioni, interrogativi e paure. Questa consapevolezza deve fortificare, accanto alla preparazione scientifica, l’empatia del Neurochirurgo che diventa non solo il medico ma il punto di riferimento, il confidente e la guida nel percorso di cura.
Presentazione clinica
I tumori cerebrali possono manifestarsi diversamente a seconda della sede della lesione e dei rapporti anatomici che essa contrae. In alcuni casi i sintomi sono sfumati e lenti a comparire; in altri si presentano in maniera più eclatante, come emorragie, crisi epilettiche, deficit neurologici conclamati. Lo spettro dei sintomi spesso è legato anche all’effetto massa ovvero alla crescita di una lesione occupante spazio e alla conseguente sofferenza delle strutture limitrofe.
A seconda della localizzazione della lesione si configurano alterazioni e disturbi specifici:
- Lobo frontale: alterazioni del comportamento come apatia, abulia, irritabilità, disinibizione; cambiamenti del carattere e della capacità di pianificare; difficoltà nel linguaggio e nell’iniziativa motoria; in alcuni casi, si presentano anche deficit motori parziali o completi.
- Lobo temporale: disturbi della sfera emotiva, alterazioni viso-spaziali e difficoltà nella comprensione verbale.
- Lobo parietale: disturbi della sensibilità e difficoltà nella percezione del proprio corpo nello spazio.
- Lobo occipitale: problemi visivi come alterazione della visione, restringimento del campo visivo, difficoltà nella percezione dei colori.
Terapie sintomatiche
Fin dalle prime fasi è indispensabile un inquadramento neurochirurgico.
A giudizio del medico si può ricorrere a terapie antiedemigene al fine di contenere i sintomi da effetto massa ed eventuali condizioni di ipertensione endocranica. Appartengono a questa categoria i corticosteroidi e selettivamente possono essere impiegati anche diuretici osmotici come il mannitolo. In presenza di un’epilessia secondaria alla crescita del tumore è fondamentale introdurre anche una terapia antiepilettica di copertura.
Diagnostica per immagini
Tra gli esami di primo livello la TC cerebrale (Tomografia assiale computerizzata) rappresenta uno strumento utile per una valutazione rapida e iniziale. L’indagine principe per la sua accuratezza e il dettaglio anatomico, tuttavia, è la Risonanza Magnetica con mezzo di contrasto (RMN) che fornisce informazioni più dettagliate circa la sede e i rapporti con le strutture limitrofe.
Una volta eseguiti gli approfondimenti diagnostici necessari il Neurochirurgo dovrà esplicitare al paziente le opzioni di trattamento, accompagnandolo nella delicata fase di comprensione della malattia e condivisione del piano terapeutico, discutendo l’eventuale spazio di un trattamento chirurgico e pianificando, ove necessario, le terapie complementari come la chemioterapia e la radioterapia.
Classificazione dei tumori cerebrali
La classificazione attualmente adottata a livello internazionale è quella definita dalla World Health Organization (WHO). Essa distingue i tumori del sistema nervoso centrale in base alle caratteristiche istologiche, cioè alla somiglianza delle cellule tumorali con le cellule normali o embrionali da cui derivano.
Tuttavia, negli ultimi anni, grazie ai progressi della biologia molecolare, la classificazione tradizionale si è arricchita di molte nozioni molecolari inerenti all’espressione di recettori e alterazioni genetiche delle cellule tumorali.
La presenza o assenza di specifici marcatori molecolari (come l’espressione di enzimi, recettori o proteine mutate) ha dimostrato di avere un ruolo determinante nella prognosi e nella risposta alle terapie, al punto da essere imprescindibile nella valutazione istopatologica e clinica.
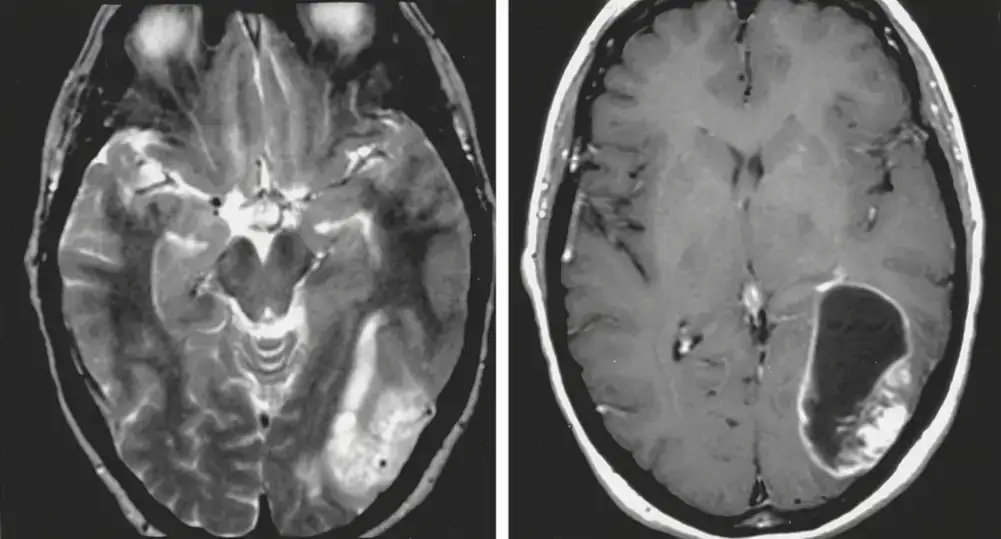
Glioma di alto grado temporale posteriore sinistro. A sinistra sequenza assiale T2 RMN. A destra sequenza RMN assiale T1 con mezzo di contrasto
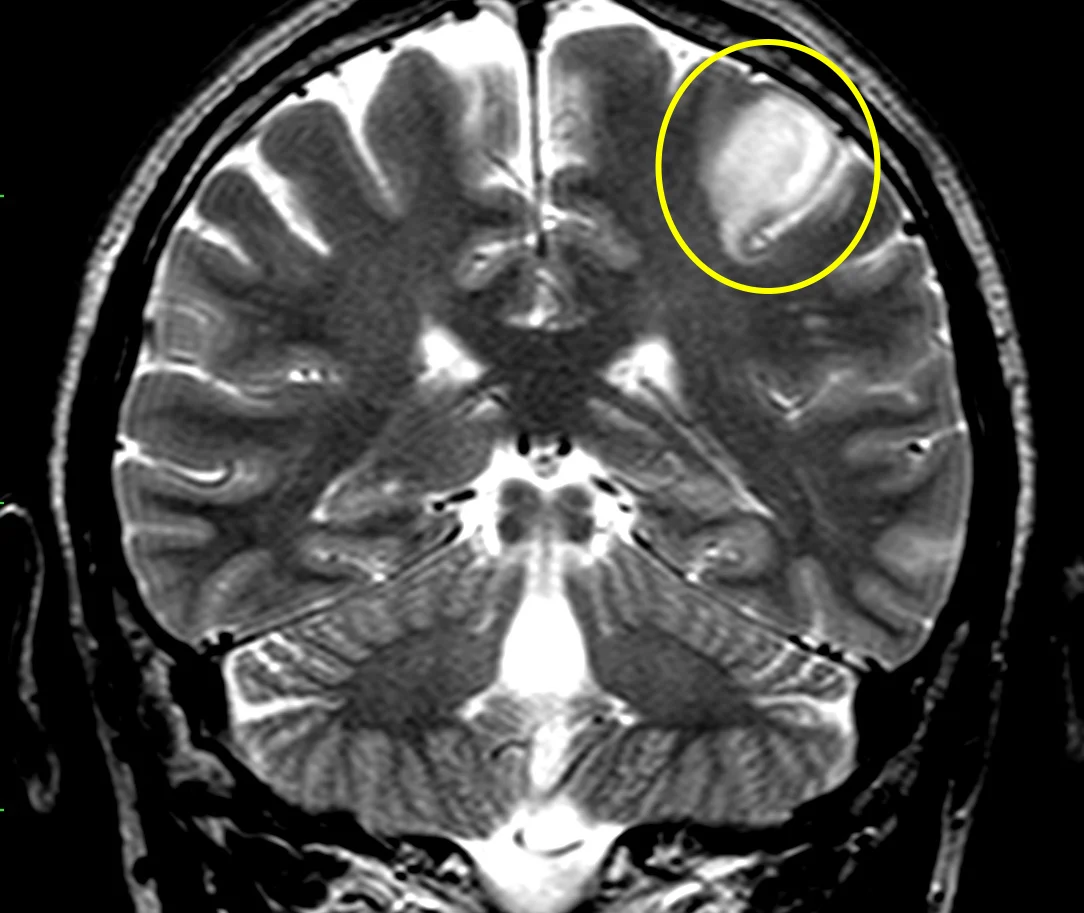
Glioma di basso grado: sequenza RMN T2, lesione cortico sottocorticale sinistra
Tumori primitivi e secondari
I tumori primitivi hanno origine dalle cellule del sistema nervoso centrale o dai tessuti di supporto (glia, meningi, plessi coroidei, guaine nervose ecc.). I tumori secondari, detti anche metastasi cerebrali, derivano invece dalla migrazione di cellule neoplastiche provenienti da altri organi.
Tumori primitivi neuroepiteliali
Questa vasta categoria include le neoplasie che originano direttamente dal tessuto nervoso o dalle sue cellule di sostegno (glia). Al suo interno si trovano:
- Astrocitomi o gliomi, che derivano dagli astrociti (cellule gliali di supporto) e si distinguono in:
- Astrocitoma pilocitico
- Astrocitoma diffuso
- Astrocitoma anaplastico
- Glioblastoma
- Oligodendrogliomi, originati dagli oligodendrociti, cellule deputate alla formazione della guaina mielinica.
- Ependimomi, che comprendono l’ependimoma classico, l’ependimoma anaplastico e il subependimoma. Derivano dalle cellule ependimali che rivestono i ventricoli cerebrali.
- Tumori del plesso coroideo, come il papilloma e il carcinoma del plesso coroideo. Queste lesioni originano dall’epitelio dei plessi, le strutture responsabili della produzione del liquido cerebrospinale.
- Tumori neuronali o misti glioneuronali, tra cui: gangliocitoma, ganglioglioma, DNET (tumore disembrioplastico neuroepiteliale), neurocitoma. Alcuni hanno origine puramente neuronale, altri presentano una composizione mista.
- Tumori della regione pineale, come pinealomi, pinealoblastomi e tumori a cellule germinali. Questi ultimi possono derivare dal tessuto della ghiandola pineale oppure da cellule germinali ectopiche presenti in quella sede.
- Tumori embrionali, tra cui il medulloblastoma e il tumore neuroectodermico primitivo (PNET).
Focus sui gliomi
Tra tutti i tumori neuroepiteliali i gliomi o astrocitomi sono tra i più comuni e rilevanti dal punto di vista epidemiologico. Con l’avvento della nuova classificazione molecolare, la descrizione istologica è accompagnata da una nuova nomenclatura molecolare a forte inferenza prognostica e terapeutica.
Le sigle molecolari più importanti includono:
- IDH (Isocitrate Dehydrogenase) mutato o wild-type
- MGMT (O6-methylguanine-DNA methyltransferase)
- Codelezione 1p/19q (tipica degli oligodendrogliomi)
- Istone H3 K27M, tipico di forme pediatriche o localizzazioni mediane profonde ad alta malignità
(Vedi anche i documenti: “Gliomi Subclassifications 2017” e “WHO Classification 2021”)
Dal punto di vista clinico, i gliomi possono presentarsi come lesioni focali a comportamento benigno – come l’astrocitoma pilocitico – oppure lesioni altamente infiltranti con una marcata tendenza alla ricorrenza e alla recidiva – come nel caso dell’astrocitoma diffuso, astrocitoma anaplastico e glioblastoma.
Differenti modalità di presentazione e predilezione mostrano anche i tumori gliali in età infantile e adulta. L’età del paziente correla infatti con il tipo di glioma e la sua localizzazione. Se negli adulti i gliomi sono tendenzialmente più aggressivi a sede emisferica, al contrario, l’astrocitoma pilocitico è tipico dell’infanzia e dell’adolescenza e si localizza più frequentemente nel cervelletto o nella regione del terzo ventricolo.
Le lesioni di natura gliale specialmente le forme di alto grado e altamente infiltranti hanno una spiccata tendenza alla disseminazione. Le vie di migrazione delle cellule tumorali includono:
- i fasci di sostanza bianca specie le formazioni commissurali (come il corpo calloso)
- la via liquorale attraverso il liquido cerebrospinale
- la diffusione ependimale (lungo i ventricoli)
- la diffusione piale (il sottile rivestimento meningeo più adeso al parenchima cerebrale)
- gli spazi perivascolari
Il “gold standard”: l’importanza della diagnosi e della chirurgia
La tempestività nella diagnosi e il corretto inquadramento clinico fin dall’esordio sono elementi indispensabili per affrontare e curare la malattia. La visita specialistica neurochirurgica e il colloquio clinico, infatti, consolidano l’esigenza di una pianificazione terapeutica che trova nella chirurgia il primo razionale. Ove possibile, la chirurgia con finalità di resezione radicale rappresenta la prima scelta di trattamento. L’obiettivo del gesto chirurgico resta sempre l’asportazione completa della lesione, declinabile, solo in casi selezionati ad un’asportazione subtotale con finalità di preservazione dell’integrità neurologica. La radicalità del gesto chirurgico ha infatti significato prognostico: per alcuni istotipi come l’astrocitoma pilocitico, la resezione completa può essere curativa. Nelle forme più invasive e infiltranti (astrocitoma diffuso, anaplastico, glioblastoma), la chirurgia di resezione rappresenta il presupposto essenziale per il successo di terapie concomitanti come la chemioterapia e la radioterapia aumentando l’impatto sulla sopravvivenza e riducendo il rischio di recidiva.
Meningiomi
Menigioma della faccia posteriore della rocca: Sequenza RMN assiale T1 con mezzo di contrasto di meningioma della faccia posteriore della rocca a destra.
A sinistra sequenza RMN assiale T1 con mezzo di contrasto di meningioma a partenza dalle docce olfattorie. A destra sequenza RMN coronale T1 con mezzo di contrasto di meningioma delle docce olfattorie.
Tra i tumori primitivi del sistema nervoso centrale si citano i tumori di derivazione meningea o meningiomi che occupano un ruolo di rilievo. Come si evince dall’accezione essi originano dalle cellule meningoteliali dell’involucro meningeo che avvolge il parenchima cerebrale (dura madre, aracnoide, pia madre) e che forma anche pliche di sostegno e separazione come la falce cerebrale, il tentorio, il planum e il dorsum sellae. Nella maggior parte dei casi si tratta di lesioni indolenti a comportamento benigno che possono essere, per sede e localizzazione, meritevoli di un trattamento chirurgico il più delle volte risolutivo.
La chirurgia rappresenta infatti il trattamento di elezione specialmente per quelle lesioni a basso indice proliferativo (Ki 67) per cui si rivela risolutiva e curativa. E’ evidente che un ruolo fondamentale riveste l’esame istologico che potrà chiarire l’esatto comportamento biologico e identificare, pertanto, quei sottotipi istologici che mostrano una maggiore tendenza alla recidiva per i quali è utile contemplare controlli post-operatori ravvicinati.
Adenomi Ipofisari
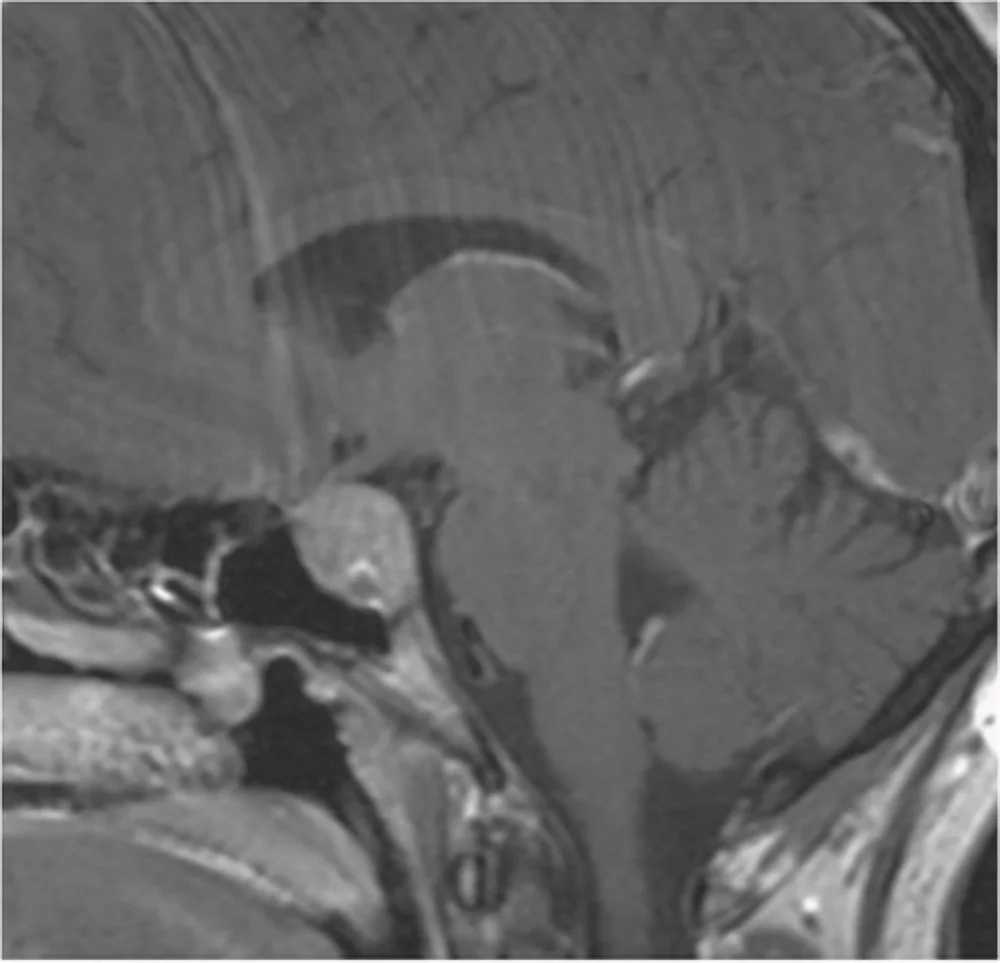
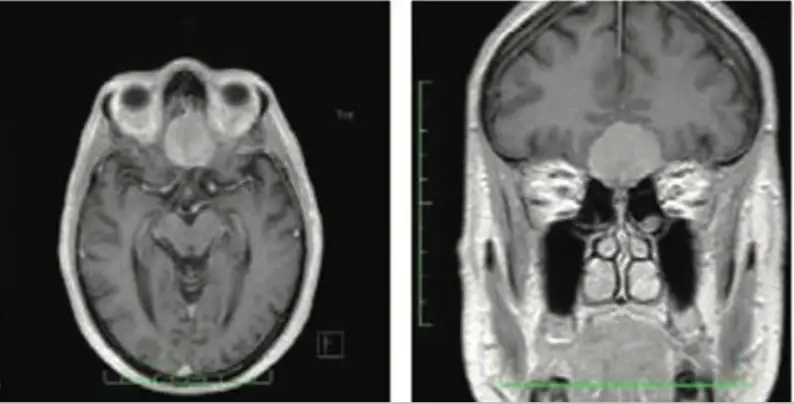
Adenoma ipofisario: sequenza sagittale RMN T1 con mezzo di contrasto, si apprezza una lesione sellare aggettante nel seno sfenoidale.
Adenoma ipofisario: sequenza coronale RMN T1 con mezzo di contrasto, la lesione sellare appare a ridosso del tratto cavernoso delle due arterie carotidi interne.
Un’altra importante categoria di tumori primitivi è rappresentata dagli adenomi ipofisari, neoplasie benigne che originano dalle cellule ipofisarie dell’omonima ghiandola localizzata nella regione sellare alla base del cranio.
Queste lesioni sono responsabili dell’insorgenza di quadri clinici tipici. Caratteristico è infatti il disturbo visivo descritto come emianopsia bitemporale ovvero la perdita della visione periferica nei quadranti temporali (laterali) di entrambi gli occhi. Spesso questo disturbo è patognomonico e deriva dalla compressione del chiasma ottico una struttura nervosa che contrae un rapporto di vicinanza con la sella turcica sede della ghiandola ipofisaria
Gli adenomi ipofisari si suddividono in secernenti e non secernenti. I primi producono in eccesso ormoni come prolattina (PRL), ormone della crescita (GH), ACTH (ormone adrenocorticotropo), TSH (ormone tireostimolante), FSH (ormone follicolostimolante), determinando squilibri endocrinologici. I secondi, invece, non alterano la secrezione ormonale o possono determinare solo squilibri ormonali da deafferentazione ipotalamica e spesso vengono diagnosticati solo in seguito a sintomi da compressione (disturbi visivi).
In base alle dimensioni, si distinguono:
- Microadenomi (<10 mm)
- Macroadenomi (>10 mm)
È fondamentale ai fini della di un corretto inquadramento diagnostico iniziale considerare anche altre alterazioni e lesioni di natura non adenomatosa che possono interessare la stessa sede anatomica (sella turcica):
- Ipofisiti (infiammatorie o autoimmuni)
- Iperplasia ipofisaria (frequente in gravidanza e post-partum)
- Apoplessia ipofisaria (emorragia improvvisa della ghiandola con necrosi)
- Cisti della tasca di Rathke (residui embrionali)
- Craniofaringiomi, che derivano anch’essi dalla tasca di Rathke e si presentano con pattern distinti:
- Sottotipo adamantinomatoso, più comune in età pediatrica e anziana
- Sottotipo papillare, più frequente nell’adulto
Neurinomi (o Schwannomi)
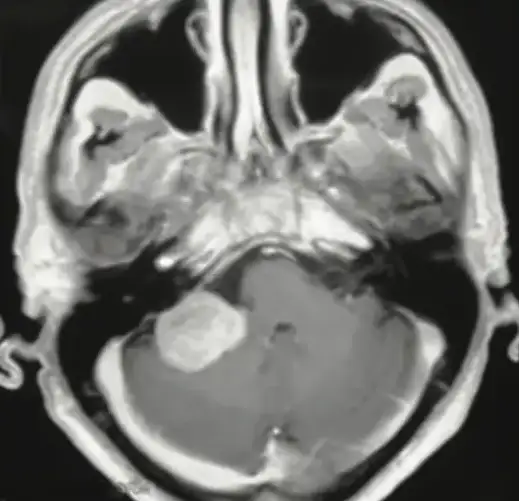
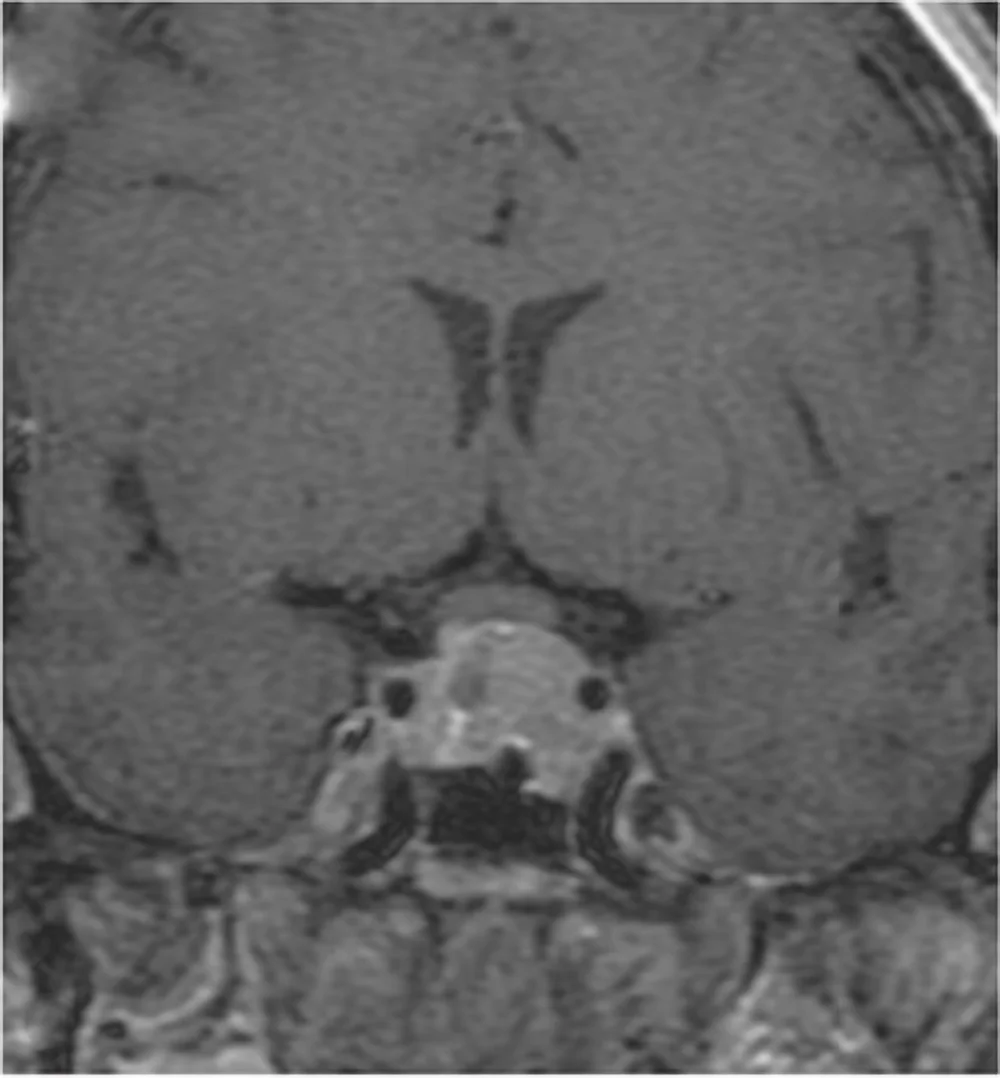
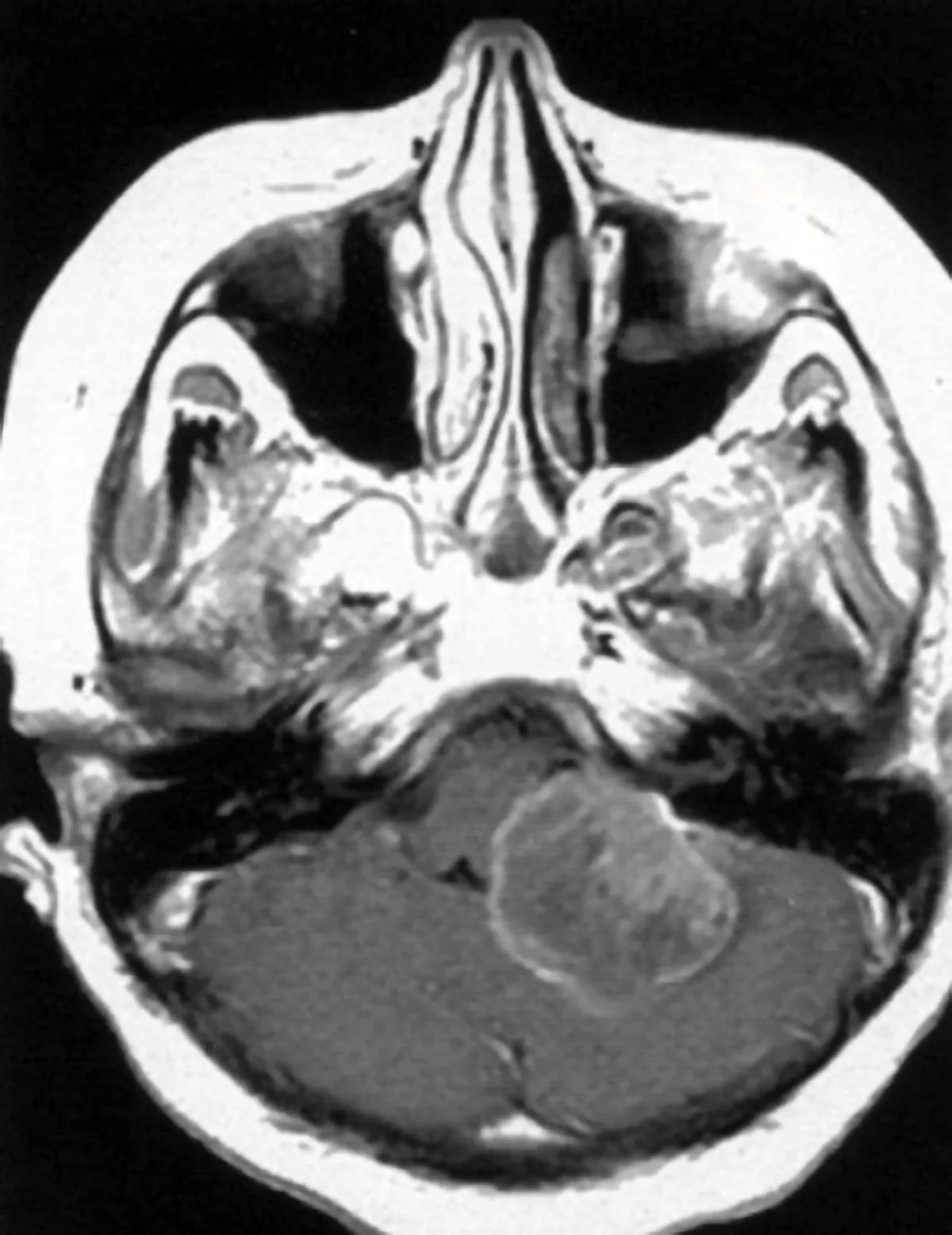
Neurinoma gigante dell’angolo ponto cerebellare destro: Sequenza RMN cisternale assiale.
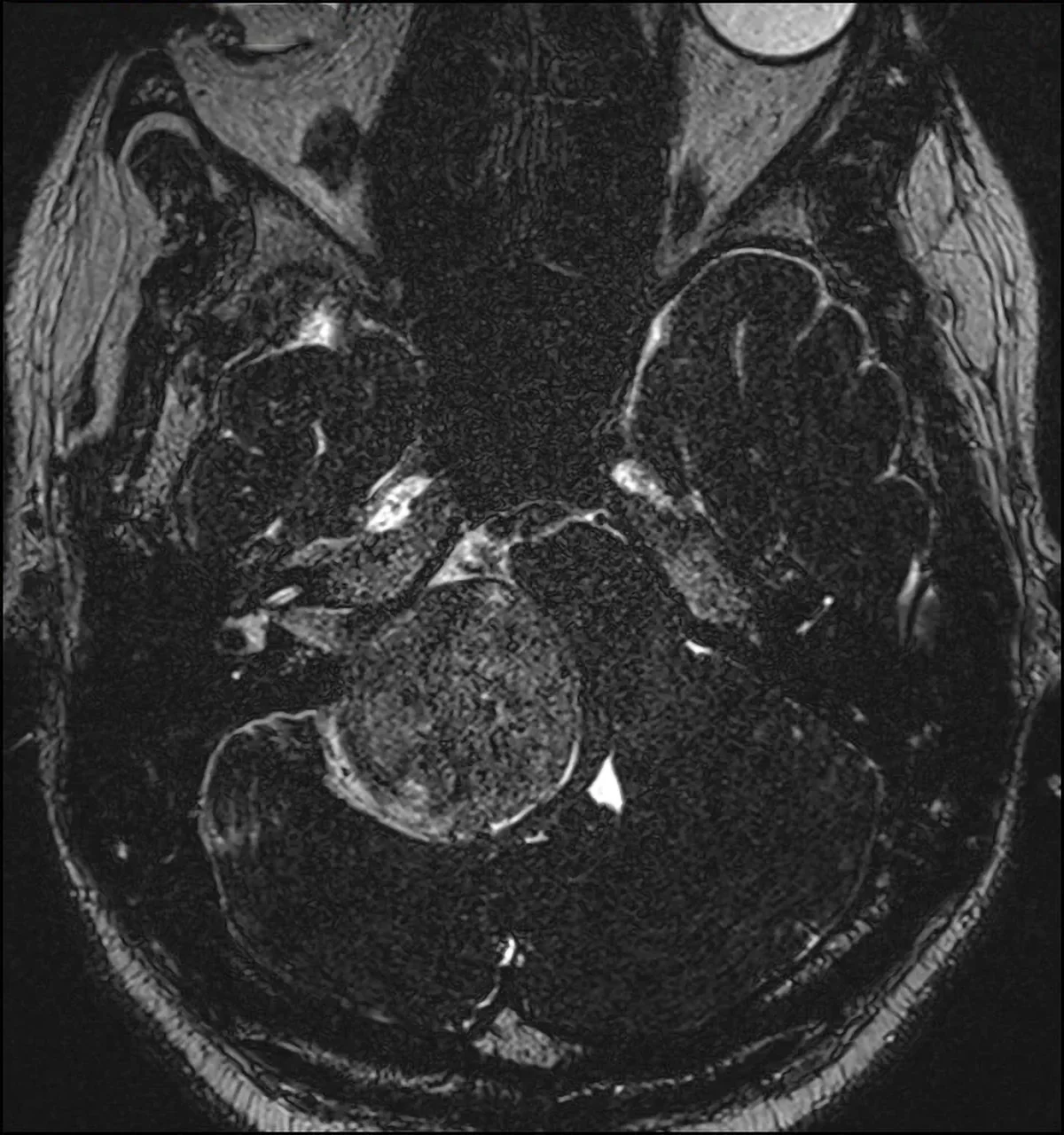
Neurinoma gigante dell’angolo ponto cerebellare destro: sequenza RMN T1 con mezzo di contrasto, caratteristica forma a “cono gelato”con estensione intra-extrameatale.
I neurinomi o schwannomi o tumori delle guaine nervose sono tumori tendenzialmente benigni che derivano dalle cellule di Schwann, responsabili della formazione della guaina mielinica dei nervi cranici e spinali. Nonostante la natura sia benigna possono mostrare comportamenti più o meno aggressivi in base al tasso di crescita (indice proliferativo) e ai rapporti anatomici che contraggono con le strutture nervose spesso ragione della comparsa dei sintomi.
Questi tumori possono comparire come forme isolate (neurinoma unilaterale singolo), oppure palesarsi nel contesto di sindromi neurocutanee come la Neurofibromatosi di tipo 1 o 2 (neurinomi multipli e bilaterali spesso associati ad altre manifestazioni e lesioni tumorali e non). Sebbene tutti i nervi cranici possano esserne colpiti, le localizzazioni più comuni interessano:
- Il III nervo cranico (oculomotore)
- Il V nervo cranico (trigemino)
- Il VII- VIII nervo cranico (complesso acustico-facciale)
Le manifestazioni cliniche variano in base al nervo coinvolto:
- Ptosi oculare nel caso del III nervo cranico
- Nevralgia facciale, ipostenia masticatoria e parestesie con coinvolgimento del V nervo cranico
- Acufeni, vertigini, ipoacusia e asimmetrie del volto in caso di interessamento del pacchetto VII-VIII nervo cranico
La chirurgia in mani esperte può essere risolutiva ma richiede estrema precisione e dovizia tecnica. Indispensabile è la pianificazione di un gesto chirurgico con l’ausilio del monitoraggio neurofisiologico intraoperatorio affinché la resezione chirurgica massimale o submassimale sia condotta a preservazione della funzione nervosa. In alcuni casi selezionati, può essere contemplata, come alternativa meno invasiva ma non scevra da rischi, la radiochirurgia stereotassica previa accurata valutazione dei rischi e benefici.
Linfoma cerebrale
Il linfoma primitivo del sistema nervoso centrale (PCNSL Primary Central Nervous System Lymphoma) è una neoplasia maligna di origine linfomatosa che origina direttamente nel sistema nervoso centrale e deve essere differenziata da eventuali altre localizzazioni cerebrali secondarie a malattie linfomatose sistemiche.
Il PCNSL può manifestarsi sia in pazienti immunocompetenti che in soggetti immunodepressi. La presentazione può avvenire sottoforma di singola lesione o lesioni multiple a localizzazione emisferica cortico-sottocorticale e in sede paraventricolare. Nella stragrande maggioranza dei casi (circa il 95%) si tratta di linfomi a cellule B.
La diagnosi precoce è fondamentale per pianificare tempestivamente il trattamento. Dal punto di vista chirurgico, il ruolo della chirurgia è limitato, generalmente circoscritto alla biopsia stereotassica o alla resezione parziale con finalità di diagnosi isto-patologica.
Il trattamento può prevedere il ricorso a:
- Chemioterapia
- Radioterapia
- Terapia cortisonica
La percentuale di risposta a queste terapie è elevata (fino al 70-80%), ma il rischio di recidiva resta concreto, imponendo una stretta sorveglianza clinica e follow up radiologico.
Metastasi cerebrali
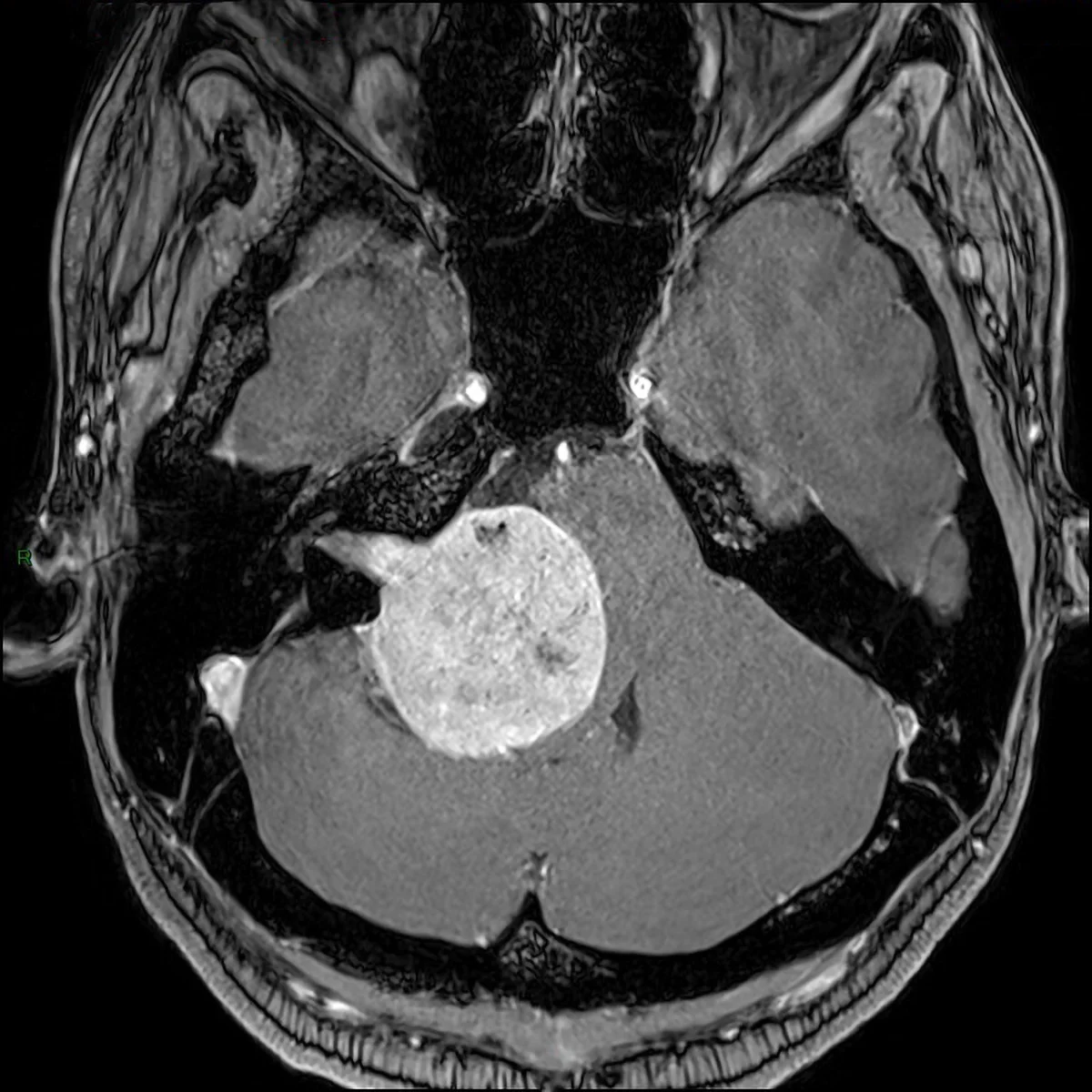
Metastasi in fossa cranica posteriore: sequenza RMN assiale T1 con mezzo di contrasto, lesione a livello del peduncolo cerebellare medio sinistro con compressione del tronco encefalico e del IV ventricolo (metastasi da carcinoma polmonare).
Il grande capitolo dei tumori secondari comprende le metastasi cerebrali che originano dalla migrazione e dalla proliferazione incontrollata di cellule neoplastiche derivanti da un tumore primario localizzato in un altro organo. La via principale di disseminazione è quella ematogena, ma in alcuni casi si possono osservare anche disseminazioni per contiguità per esempio attraverso le guaine nervose o i vasi in corrispondenza di forami ossei e fessure craniche.
La crescita di una metastasi cerebrale non è un semplice processo proliferativo. Si tratta di un fenomeno complesso biologicamente e geneticamente regolato, in cui cellule tumorali migranti riescono non solo a colonizzare il cervello, ma anche a creare un microambiente favorevole alla loro sopravvivenza e proliferazione. Questo avviene attraverso:
- L’attivazione di proto-oncogeni
- L’inibizione di geni oncosoppressori
- L’angiogenesi tumorale con la creazione di un microambiente vascolarizzato che garantisce l’adeguato supporto metabolico
Nel 10% dei casi la diagnosi di metastasi cerebrale costituisce il primo evento in quanto non è ancora noto il tumore primitivo.
Nel 10% dei casi, il cervello è l’unico organo colpito.
Nell’80% dei casi la localizzazione è intraparenchimale emisferica, seguita dall’osso cranico e dalla dura madre (15%).
L’interessamento leptomeningeo e la carcinomatosi meningea risultano eventi più singolari e rari (5%).
Quanto al numero di lesioni:
- Nel 50% dei casi la metastasi è singola
- Nel 30% sono presenti più di tre lesioni
- Nel 5% si osservano oltre cinque lesioni
I tumori primitivi più frequentemente responsabili di metastasi cerebrali sono:
- Carcinoma mammario
- Carcinoma polmonare
- Tumore renale
- Carcinoma del colon
- Melanoma
Trattamento delle metastasi cerebrali
L’intervento neurochirurgico trova indicazione solo in presenza di una metastasi solitaria o di lesioni adiacenti accessibili e aggredibili chirurgicamente. Va ricordato, però, che se la chirurgia trova il razionale nell’exeresi radicale della lesione, essa non esercita alcun controllo sulla malattia primitiva all’origine della disseminazione di cellule neoplastiche. Per questo motivo il risultato chirurgico richiede l’intervento cooperativo di terapie adiuvanti come la radioterapia e la chemioterapia.
Un approccio multidisciplinare è sempre essenziale per la gestione ottimale di questi pazienti.